
에피스 스토리
에피스 스토리
영문으로 운영되고 있는 삼성바이오에피스 SNS에 포스팅된 글입니다.

[Re:Access] Biosimilar Regulation is Improving, But Still Needs Work
Welcome back to Re:Access, a newsletter series by Samsung Bioepis that Rethinks biosimilars as the answer to future healthcare Access.
In our third edition, we consider what regulatory milestones have had a significant impact on biosimilar development in recent years, and how much more needs to be done?
Let’s dive in.
What Regulatory Hurdles Do Biosimilar Manufacturers Face?
Right at the end of last month, October 2025, the U.S. Food and Drug Administration (FDA) published a new draft guidance. In it, they propose major updates to streamline the process of evaluating whether there are clinically meaningful differences between the proposed biosimilar and the reference product in terms of safety, purity, and potency. It’s a significant step forward in regulatory streamlining, following the combined efforts of global stakeholders championing science over routine.
As we’ve highlighted in previous issues, burdensome regulatory requirements are one of the reasons behind the lack of biosimilars in development. Looking back, we can track how far the regulatory environment has come since the first biosimilar was introduced to the market.
When biosimilars were relatively new to the biologics space in the US, the Biologics Price Competition and Innovation Act (BPCIA) of 2010 created a regulatory pathway for the approval of biosimilars. Based on the “totality of evidence” approach, this pathway required clinical comparative efficacy studies (CES) to confirm that biosimilars were as safe and effective as their reference products.
As biosimilars became more widely used in the years following, however, it became clear that while CES can effectively prove the efficacy and safety of the original biologics, such studies do not add extra scientific value when it comes to biosimilars. This is because these aspects are already proven for the reference product.
So, what do more efficient regulatory pathways look like?
How Regulatory Streamlining Impacts Biosimilar Development
Streamlining biosimilar development does not change the quality, safety, and efficacy of the biosimilar that is approved, and significantly lowers the time and cost of development.
[Graphs sourced from: Samsung Bioepis Second Quarter 2025 Biosimilar Market Report]
Impact on Average Biosimilar Development Cost and Duration
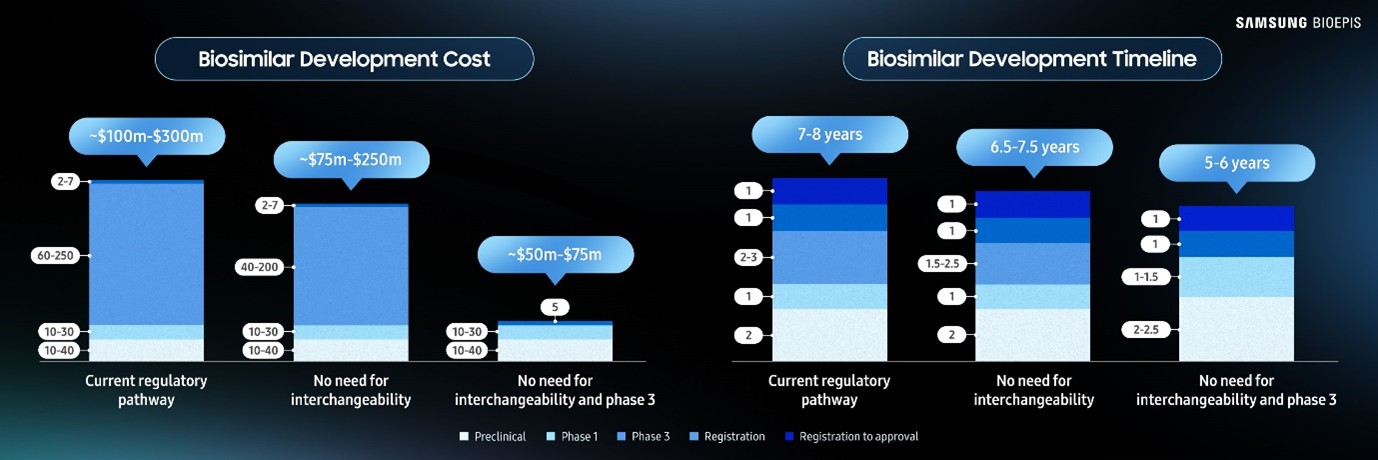
* An interchangeability designation is a legal distinction in the US allowing pharmacists to substitute the biosimilar for its reference medicine. It must be demonstrated that there is no additional risk to patients switching.
Streamlining biosimilar development means using science-based analytics. Analytical (physicochemical and functional) and clinical pharmacokinetics (PK) data alone suffices, justifying the elimination of CES besides exceptional cases where safety uncertainties cannot be resolved without additional patient exposure.
“As the Declaration of Helsinki states, we shouldn’t be ever experimenting on human beings if we’re not getting regulatorily relevant data that is the basis of the approval,” says Gillian Woollett, VP and Head of Regulatory Strategy and Policy at Samsung Bioepis. “Those analytics and increasingly functional assays, such as binding assays in vitro, are way more sensible than any feasible clinical comparative study in actual people.”
An Improved Regulatory Environment Means Better Predictability
We are observing a shift among regulatory agencies toward a more streamlined registration pathway for biosimilars. As a result, agencies are working to reduce or remove these trials from the biosimilar approval process.
Global Regulatory Streamlining in Biosimilar Development
○ 2019:FDA issues guidance, Considerations in Demonstrating Interchangeability with a Reference Product, that requires switching studies
○ 2020:Medicines and Healthcare products Regulatory Agency (MHRA) argues that in most cases, CES are not necessary and science-based analytics suffice
○ 2022:World Health Organization (WHO) updates guidelines to streamline clinical efficacy and safety comparability requirements
○ 2023:Peer-reviewed publications investigate the value of CES:
* A Data Driven Approach to Support Tailored Clinical Programs for Biosimilar Monoclonal Antibodies (E. Guillen et al)
* Do the Outcomes of Clinical Efficacy Trials Matter in Regulatory Decision-Making for Biosimilars? (N. Kirsch-Stefan et al)
○ 2024:FDA updates guidance on interchangeability to allow use of comparative analytical and clinical data instead of switching studies
○ 2025:
March:The European Medical Association (EMA) publishes Reflection Paper on a Tailored Clinical Approach in Biosimilar Development
May:International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) introduces Framework for Determining Utility of Comparative Efficacy Studies in Biosimilar Development Programs — a new ICH Multidisciplinary Guideline to address the utility of CES in biosimilar development
October:FDA publishes draft guidance, Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Updated Recommendations for Assessing the Need for Comparative Efficacy Studies, to simplify biosimilarity studies and reduce unnecessary clinical testing
We can expect the ICH guideline to encourage international regulatory bodies to reduce or eliminate CES for more streamlined development on a global scale.
“ICH is this global organization that fosters harmonized regulations around the world. So, if you get an ICH guideline, then everybody’s doing the same sorts of studies to get the same approvals,” Gillian explains. “If you’re approved in the US, you can be confident you’re approved in Europe. So, it’s harmonizing these norms so you don’t have to do separate studies in the US or separate studies in Europe.”
Streamlining Aspects Beyond Clinical Biosimilar Development
Regulatory bodies around the world are steadily increasing the efficiency of biosimilar development in their respective regions, according to the International Pharmaceutical Regulators Programme Report 2024. These steps are heartening for the industry at large, but as Gillian puts it: “The clinical trials are a particular focus, but in fact, you want all aspects to be more efficient.”
She adds, “Harmonizing regulations has value for market predictability, for commercial predictability, and development of the product. The real value of it to the industry is that they know what they’ve got to do. You get a product approved, you get access. And it’s sort of that simple, really.”
Read more about our take on recent regulatory shifts surrounding biosimilar development here.



